Assembly quality assessment#
Method#
To compare contig 1 of my assembly with the genome of the paper, QUAST was used to generate statistics report to evaluate the quality, and MUMmer was used to generated alignment dotplot to visualize the similarity.
QUAST#
Command:
quast.py \
-o ~/genome_analysis/analyses/02_assembly_quality_assessment/01-quast \
-r ~/genome_analysis/data/reference/OBMB01.fasta -x 700000 \
~/genome_analysis/analyses/01_genome_assembly/01-lfts-pacbio/lfts.contig1.fasta
-o: output directory
-r: reference genome
-x: lower threshold for the relocation size
MUMmer#
Command:
mummer -b \
~/genome_analysis/data/reference/OBMB01.fasta \
~/genome_analysis/analyses/01_genome_assembly/01-lfts-pacbio/lfts.contig1.fasta \
> ~/genome_analysis/analyses/02_assembly_quality_assessment/02-mummer/output.mums
-b: compute both forward and reverse complement matches
cd ~/genome_analysis/analyses/02_assembly_quality_assessment/02-mummer/
mummerplot \
-Q ~/genome_analysis/analyses/01_genome_assembly/01-lfts-pacbio/lfts.contig1.fasta \
--png \
~/genome_analysis/analyses/02_assembly_quality_assessment/02-mummer/output.mums
–png: set the output terminal to png
Results and discussion#
The report of QUAST shows the assembly is of high quality. The GC content of my assembly is 54.22%, and the one of the paper is 54.19%.
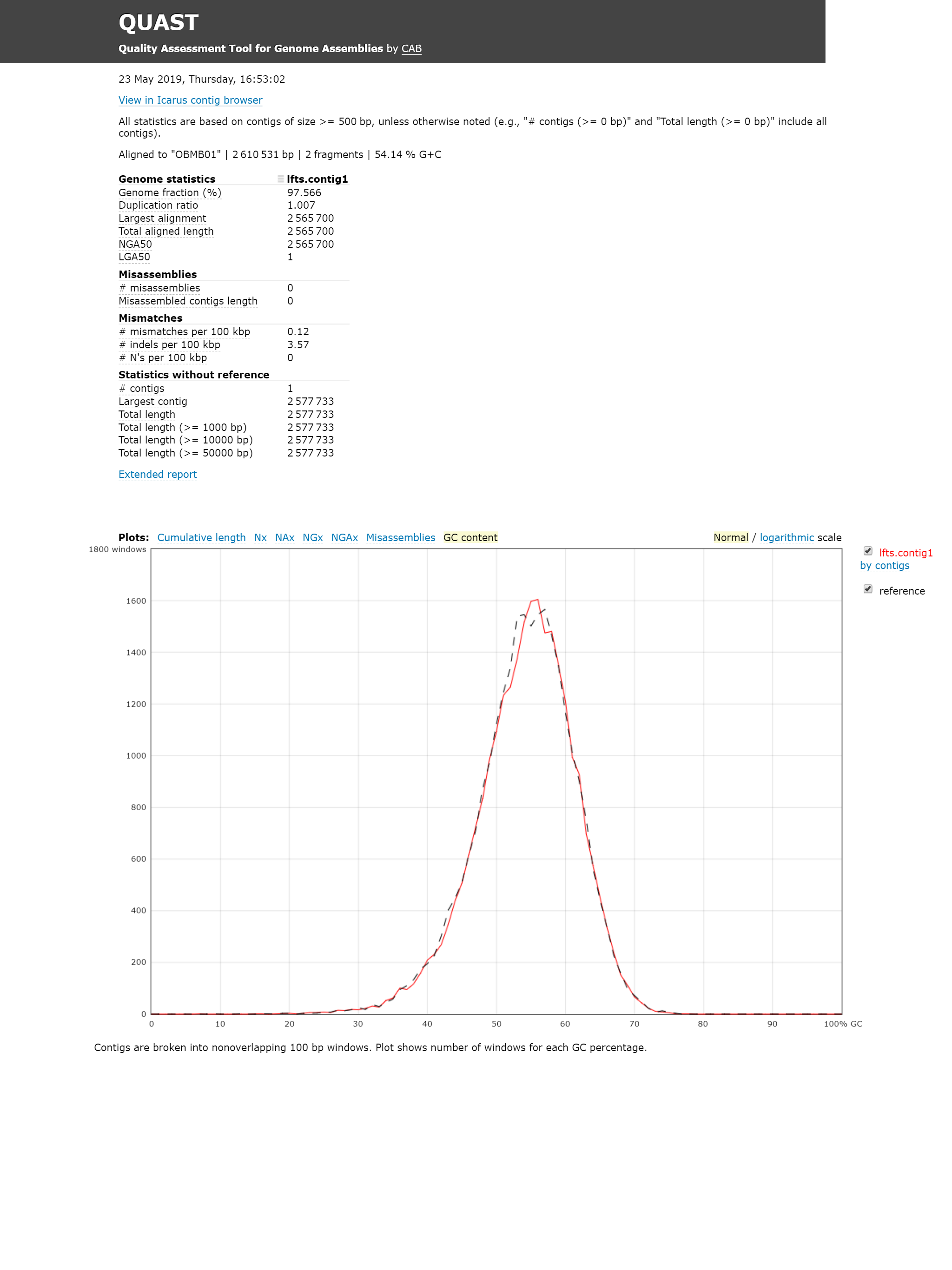
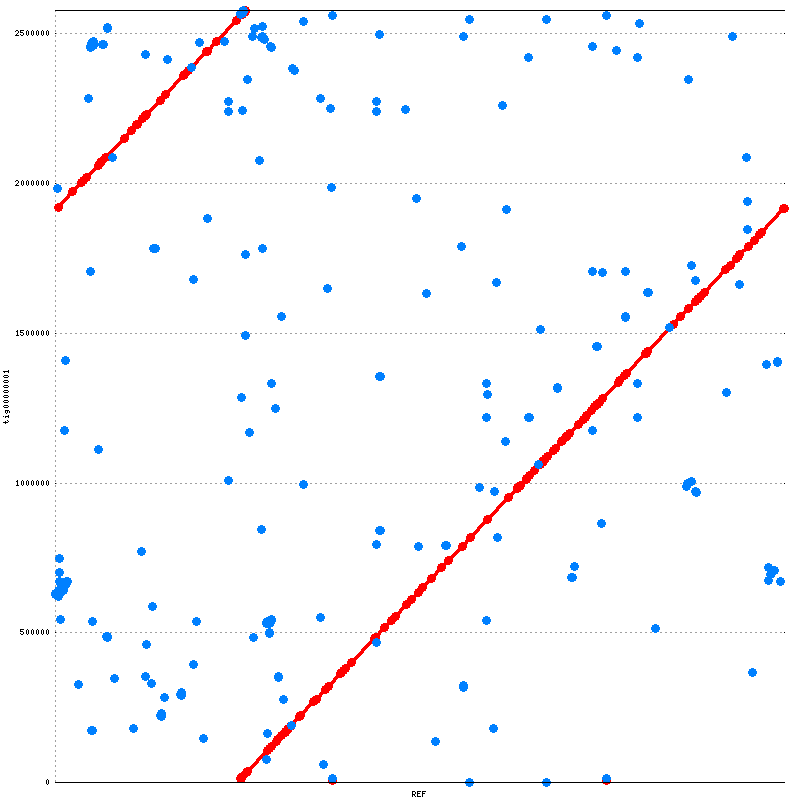 In the dotplot, the forward and reverse matches are displayed red and blue respectively. Perfect alignment will display as a red diagonal. Typically, the pattern in the graph indicates relocation. However, since prokaryotic genome is circular, and the beginning can be selected arbitrarily, these two assemblies are consistent with each other.
In the dotplot, the forward and reverse matches are displayed red and blue respectively. Perfect alignment will display as a red diagonal. Typically, the pattern in the graph indicates relocation. However, since prokaryotic genome is circular, and the beginning can be selected arbitrarily, these two assemblies are consistent with each other.
Q & A#
Assembly evaluation#
What do measures like N50, N90, etc. mean? How can they help you evaluate the quality of your assembly? Which measure is the best to summarize the quality of the assembly (N50, number of ORFs, completeness, total size, longest contig …)
N50/N90 is the sequence length of the shortest contig at 50%/90% of the total genome length. Generally, large N50/N90 suggests good quality, but other metrics should also be considered to compare results from different tools. There is not a magic rule of thumb to select the best assembly.
How does your assembly compare with the reference assembly? What can have caused the differences?
See results and discussion.
Why do you think your assembly is better/worse than the public one?
My assembly is comparable to the public one. Since prokaryotic genome is much simpler than eukaryotic, results are very similar although using different assembly software.